Research
The convergence of AI and engineering biology is redefining how we approach complex genetic diseases such as lysosomal storage disorders (LSDs). These rare, devastating conditions—driven by enzyme deficiencies that cause toxic metabolite buildup—have have remained difficult to cure through conventional drug discovery pipelines. Now, using AI, we can design optimized biotherapeutics with unprecedented precision, and build new therapies, ranging from custom enzyme variants to cell-therapies, that can restore lysosomal function, Together, these technologies are moving us from symptom management to true molecular repair. By integrating data-driven insights with wet-lab validation, researchers can iterate faster, reduce experimental uncertainty, and personalize interventions to each patient’s molecular signature. The result is an emerging paradigm where machine learning does not just guide discovery—it becomes an active collaborator in engineering biology. For lysosomal storage disorders, this synergy offers real hope: scalable, precise, and potentially curative therapies for all patients.
Our mission
Developing next generation therapies for Lysosomal Storage Diseases
We are building on our expertise in AI and synthetic genomics to design and build a next-generation of enzyme replacement therapies. Our goal is to establish technologies to optimise the therapeutic properties of recombinant enzymes and to engineer expression systems for inexpensive production of these molecules, in order to provide new sustainable treatment for patients with lysosomal storage diseases, particularly focusing on Fabry disease.

Our technologies
Generative Artificial Intelligence for biologics engineering

Our backbone framework are variational autoencoders, which we optimise for training and design efficiency using our experience built across 20 years of statistical learning, optimisation and biological data analysis methods development.
Our methods build on robust engineering frameworks and industry standards, such as Python, Git, GitHub and GPU computing, and deployed using the Nextflow workflow management systems.
Genome engineering for biologics production
We are now building on our expertise in synthetic genomics and engineering biology to engineering microbial and mammalian cells to optimise biologics production. Our workhorse systems are Komagathella Phaffi (aka P. Pastoris) and Chinese Hamster Ovary (CHO) cell lines, primarily engineered to efficiently produce human lysosomal enzymes.

Automated high-throughput biology
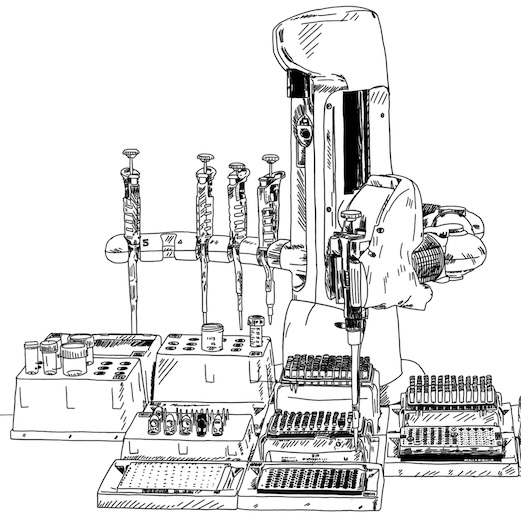
To make sure our experimental workflow keeps up with our generative AI models, we are developing miniaturized and fully automated protocols for protein expression in both Komagathella Phaffi (aka P. Pastoris) and Chinese Hamster Ovary (CHO), using high-throughput electroporation systems, liquid handling robots, and highly throughput assays.
Our lab works in close collaboration with the Edinburgh Genome Foundry to scale-up our experimental work.
Other areas of interest
Cancer genetics and genomics
Funding
![]()
![]()
![]()
![]()
![]()
![]()
![]()